Câncer Gástrico Hereditário
Autores: Marcus Fernando Kodama Pertille Ramos e Ítalo Beltrão Pereira Simões
A maioria dos casos de câncer gástrico (CG) são esporádicos, mas cerca de 10% apresentam agregação familiar e 1 a 3% possuem uma causa hereditária. O conhecimento das síndromes hereditárias como fator causal do câncer colorretal (CCR) é bem difundido, mas no CG isso é menos divulgado, fato que pode prejudicar o diagnóstico precoce e o seguimento adequado.
O CG hereditário pode ocorrer associado a presenças de polipose ou não, como no caso das síndromes do Câncer Gástrico Difuso Hereditário, Li-Fraumeni, BRCA1, BRCA2 e Lynch. Nesse artigo descreveremos brevemente as principais síndromes de CG hereditário associadas à polipose deixando as não associadas para um artigo futuro.
1. CG HEREDITÁRIO ASSOCIADO A POLIPOSE
1.1 POLIPOSE ADENOMATOSA FAMILIAR (PAF)
A PAF decorre de uma mutação do gene supressor tumoral do gene APC causando um altíssimo risco de CCR.
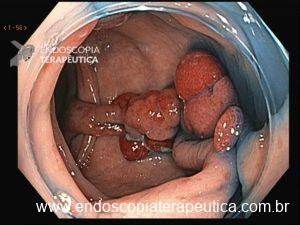
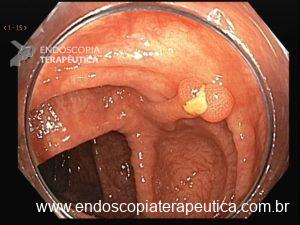
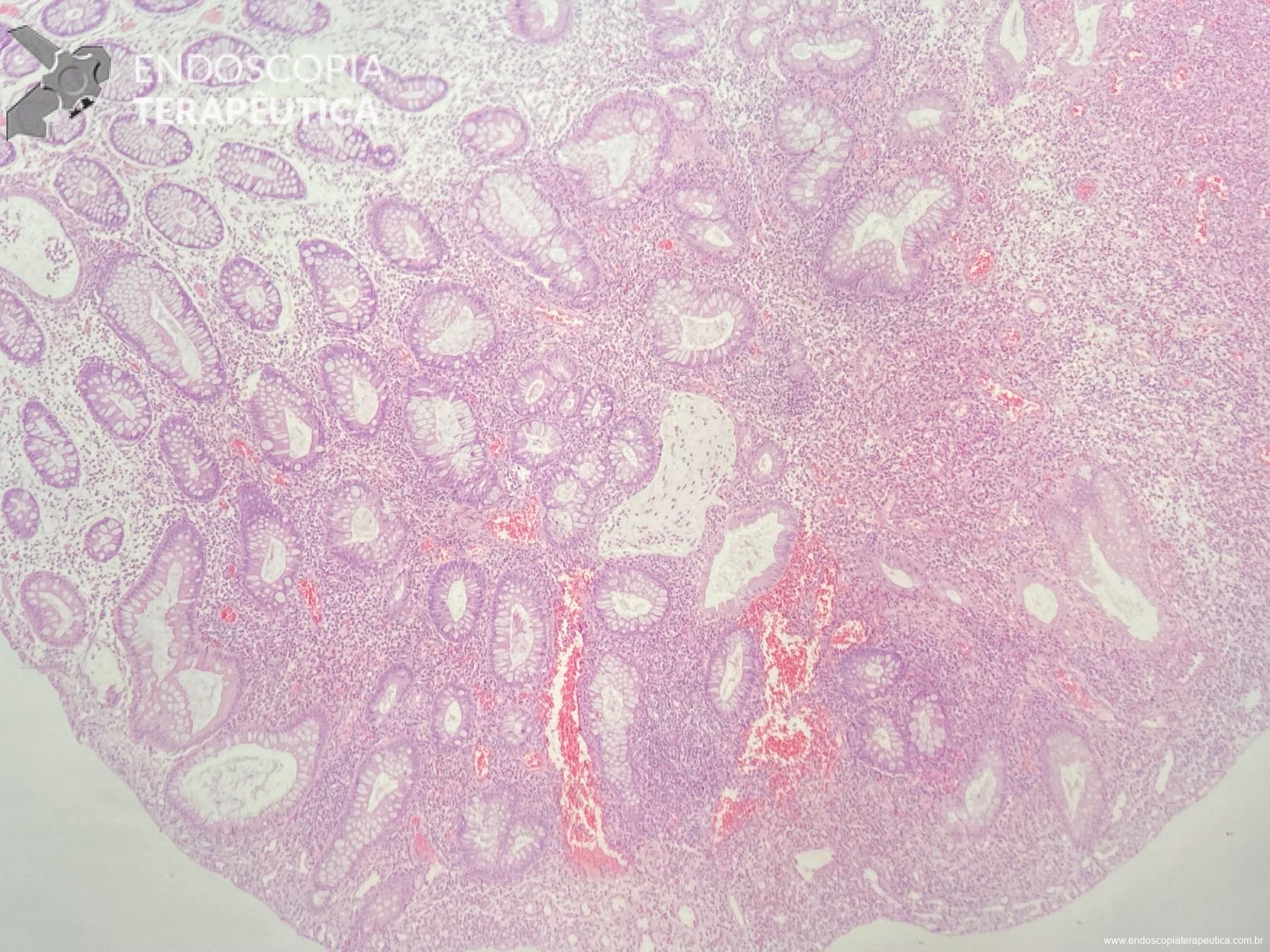
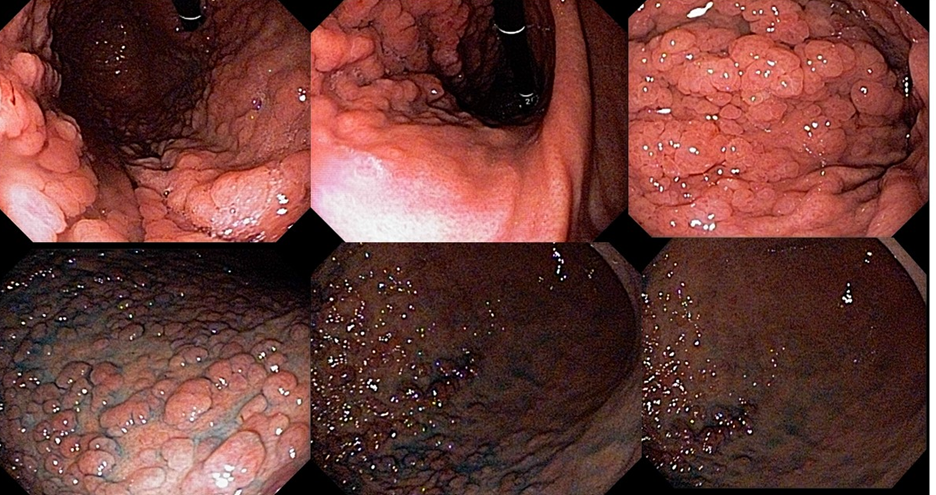
Cerca de 51 a 88% dos pacientes apresentam pólipos gástricos principalmente de glândulas fúndicas (PGF). A incidência é elevada inclusiva na PAF atenuada. Eles costumam ser numerosos, e o termo polipose gástrica pode ser empregado apenas quando mais de 20 estão presentes.
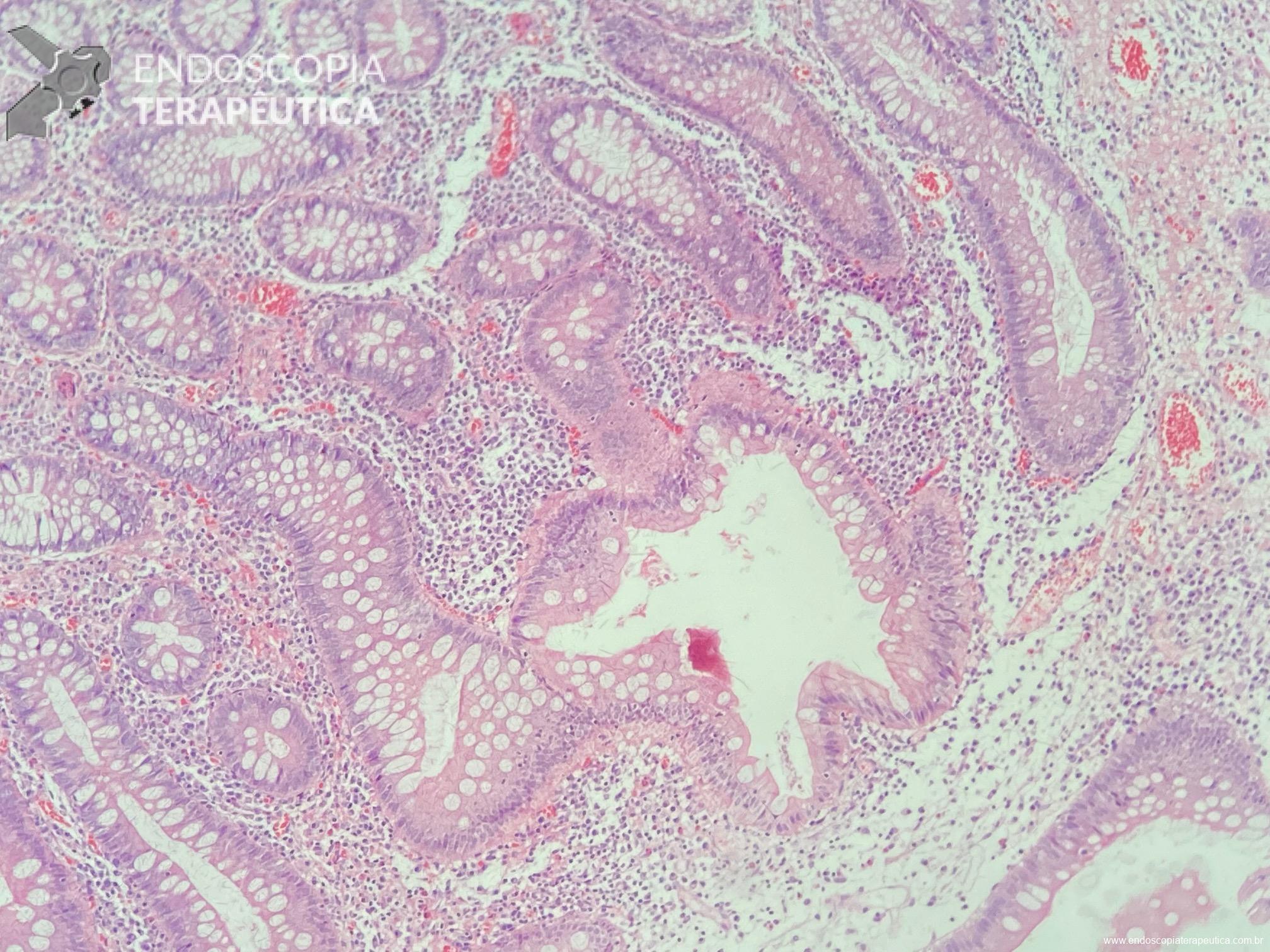
Displasia de baixo grau pode estar presente em até 44% dos pólipos de glândulas fúndicas. Pólipos adenomatosos são detectados em cerca de 20% dos pacientes com PAF.
O rastreamento endoscópico alto é recomendado no momento da manifestação da polipose colônica ou a partir de 25 anos. O intervalo de realização vai depender dos achados e também de acordo com a necessidade de seguimento de adenoma de papila, quando presente, de acordo como escore de Spigelman.
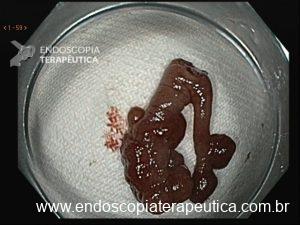
1.2 SÍNDROME DE PEUTZ-JEGHERS (SPJ)
A SPJ é autossômica dominante, caracterizada pelo desenvolvimento de polipose hamartomatosa gastrointestinal principalmente no jejuno associada a presença de máculas melanocíticas.
O diagnóstico clínico baseia-se na confirmação da presença de pólipos hamartomatosos associado com história familiar positiva e hiperpigmentação de mucosas, dedos e genitália externa.
Pólipos gástricos são detectados em 25% dos casos, comparados com 70-90 % encontrados no intestino delgado e 50% no cólon. O aspecto morfológico do pólipo gástrico na SPJ assemelha-se a um padrão viloso proliferação epitelial hiperplásica sendo difícil distinguir do pólipo juvenil e hiperplásico.
Displasia é raramente detectada nos pólipos mas indivíduos com SPJ tem 29% risco de desenvolver CG principalmente do tipo intestinal.
Rastreamento deve ser iniciado precocemente na infância com endoscopia inicial com periodicidade dependendo dos achados. A partir dos 50 anos, o risco de CG aumenta e a periodicidade deve ser mais frequente entre 1 a 2 anos.
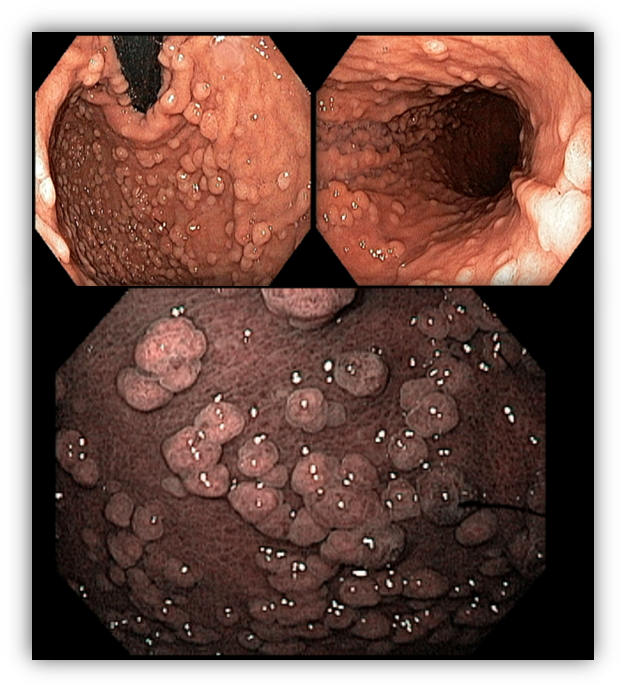
1.3 SÍNDROME DA POLIPOSE JUVENIL
Síndrome autossômica dominante que leva ao desenvolvimento de pólipos em todo trato gastrointestinal principalmente no cólon e reto.
Critérios para suspeita clínica da síndrome incluem mais de 5 pólipos juvenis colorretais, pólipos juvenis ao longo do trato gastrointestinal ou mais de 1 pólipo juvenil com história familiar positiva. O diagnóstico definitivo é realizado de um dos critérios de suspeita clínica na presença dos genes BMPR1A e SMAD4 no teste genético.
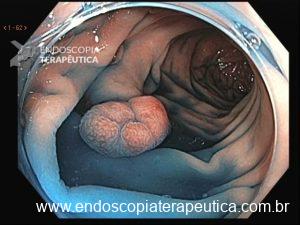
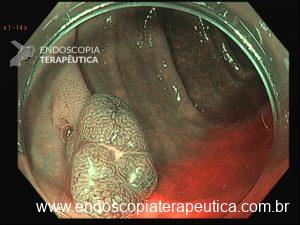
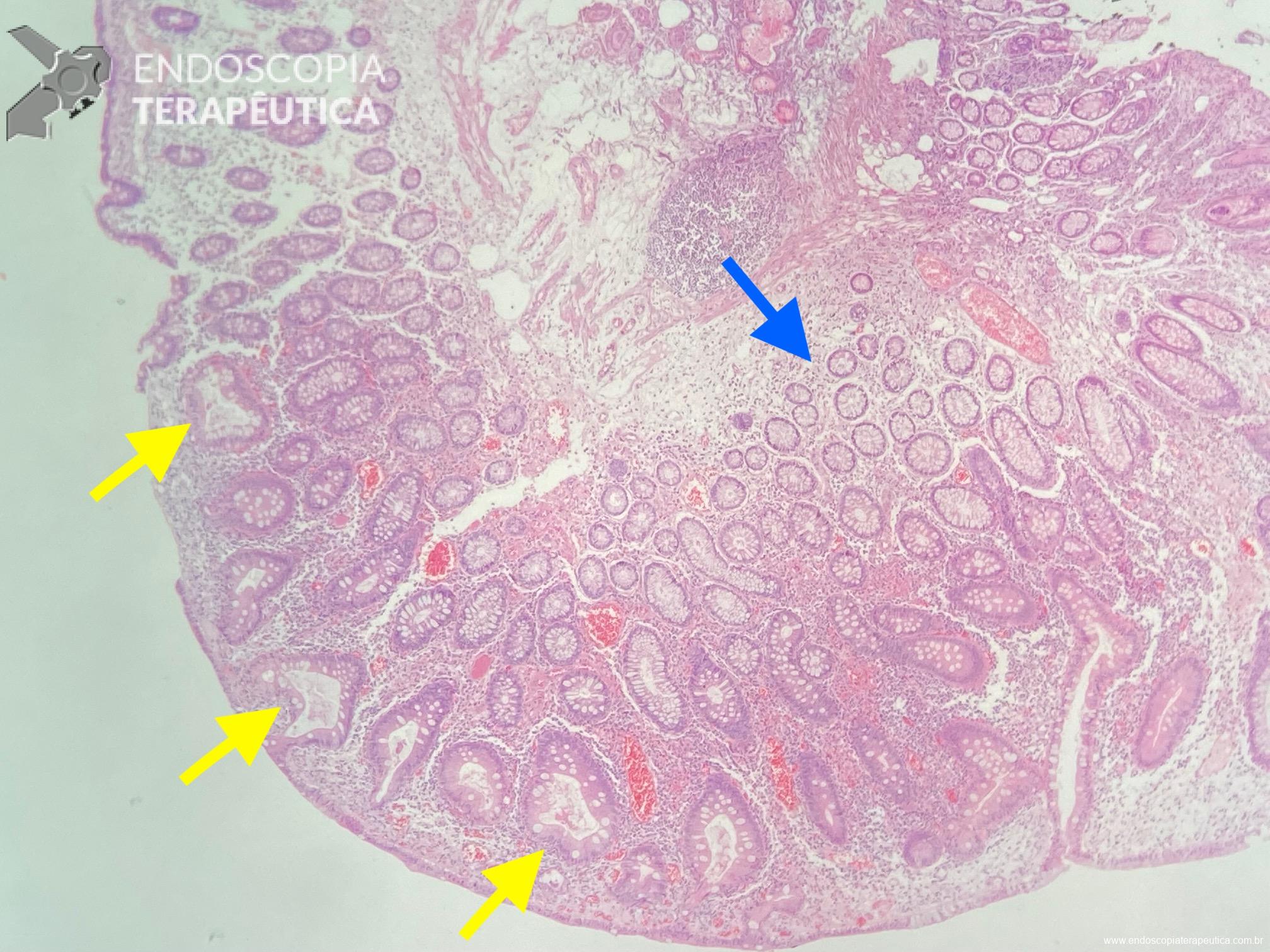
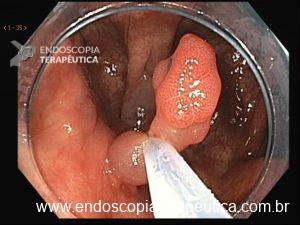
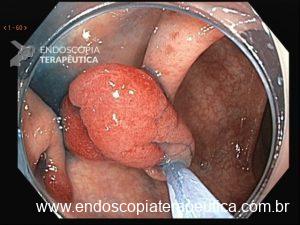
Os pólipos juvenis são pólipos hamartomatosos que se desenvolvem a partir de um tecido normal do trato gastrointestinal. O aspecto endoscópico habitualmente é de um pólipo pediculado, multilobado, macio variando desde pequenos pólipos até pólipos gigantes. Em até 75% dos casos existem outros tipos de pólipos em conjunto. Polipose gástrica severa pode ocorrer causando anemia, hematêmese, enteropatia com perda proteica e sintomas obstrutivos. Progressão para CG ocorre em até 21% dos casos com média de idade de 58 anos.
Rastreamento endoscópico é recomendado a partir da adolescência com endoscopias anuais.
Saiba mais sobre a polipose juvenil clicando neste post
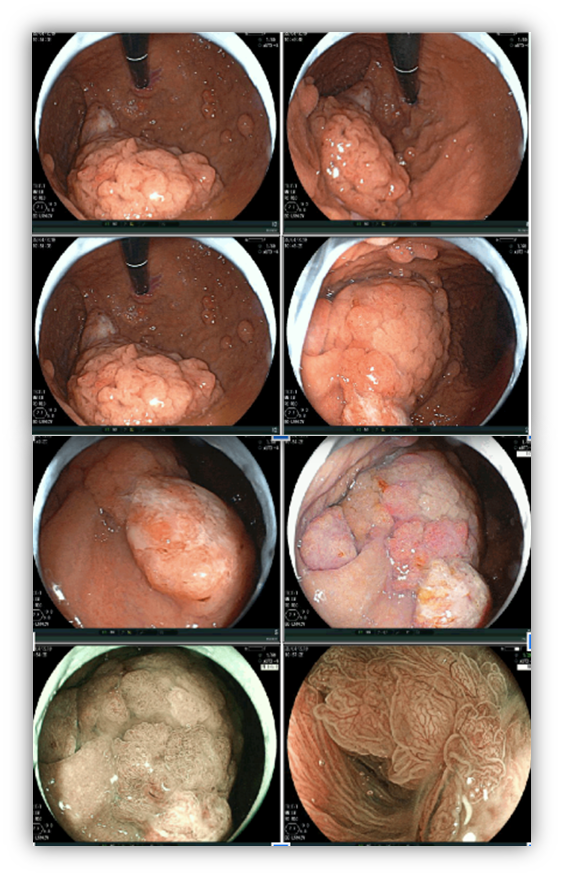
1.4 POLIPOSE ASSOCIADA AO MUTYH (MAP)
MAP é uma síndrome rara, autossômica recessiva, associada com mutação no gene MUTYH que participa de processos de reparo de DNA. Pacientes com MAP tem predisposição para o CCR, mama e ovário.
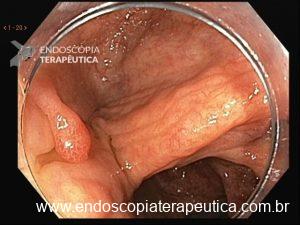
Pólipos gástricos são detectados em cerca de 10 a 33% dos casos e a maioria adenomas e PGF.
O risco de CG é baixo (2%) mas ocorre em pacientes mais jovens (mediana de 38 anos). Por outro lado, o risco de câncer duodenal é alto podendo ocorrer em 17% dos casos.
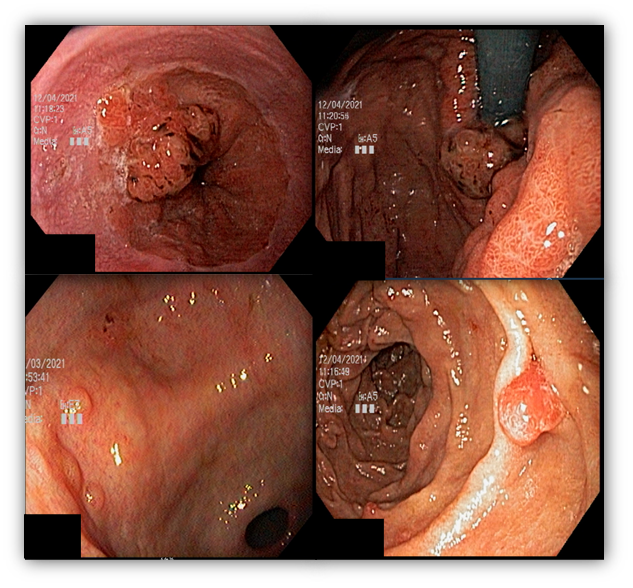
1.5 ADENOCARCINOMA GÁSTRICO COM POLIPOSE PROXIMAL (GAPPS)
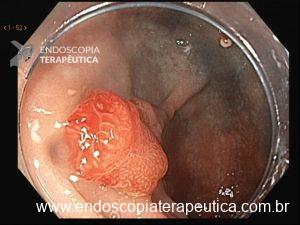
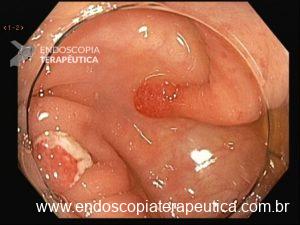
Essa síndrome é caracterizada pelo desenvolvimento de uma polipose gástrica proximal incluindo o fundo e corpo formando um tapete de pequenos pólipos usualmente menores que 1 cm. O tipo histológico dos pólipos é variado podendo ser PGF, hiperplásico, adenomas e mistos.
Os critérios para diagnóstico clínico incluem detecção de mais de 100 pólipos ou mais de 30 pólipos com história familiar positiva em parente de primeiro grau, pólipos restritos ao corpo e fundo sem a presença de pólipos colorretais, morfologia de PGF com áreas de displasia ou carcinoma, exclusão de outras síndromes e uso de inibidor de bomba de prótons.
Série de casos relataram incidência de 12,7% de CG, todos do tipo intestinal.
Seguimento endoscópico deve ser realizado, mas em casos com múltiplos pólipos a avaliação de pólipos com sinais de degeneração pode ficar prejudicada sendo indicada a gastrectomia total.

Para saber mais sobre este tema e outros relacionados, acesse o site Gastropedia clicando aqui!
Referências
- Clauditz TS, Moore M, Setia N, et al. Syndromic gastric polyposis and hereditary gastric cancers. Diagnostic Histopathologic 2019; 26(1):39-46.
- Mahon SM. Hereditary Polyposis Syndromes. Gentics and Genomics 2018; 22(2): 151-6
- Cardoso DM. Síndromes de polipose colorretal. Endoscopia Terapêutica; 2020. Disponível em: https://endoscopiaterapeutica.net/pt/assuntosgerais/sindromes-de-polipose-colorretal/
Como citar este artigo
Kodama, MFKP e Simoes IBP. Câncer gástrico hereditário. Gastropedia 2022. Disponível em: https://endoscopiaterapeutica.net/pt/assuntosgerais/cancer-gastrico-hereditario