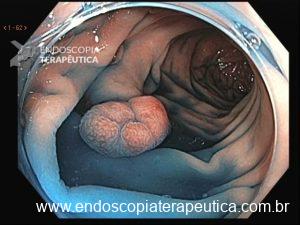
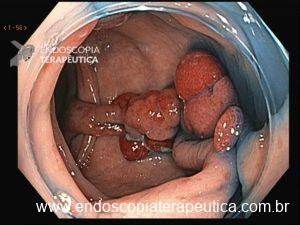
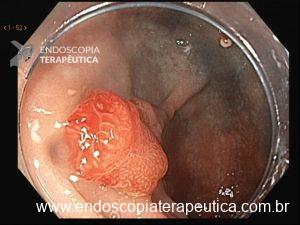
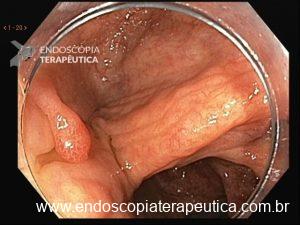
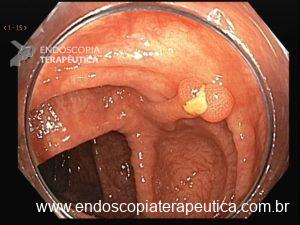
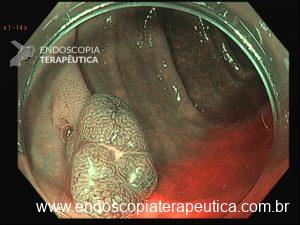
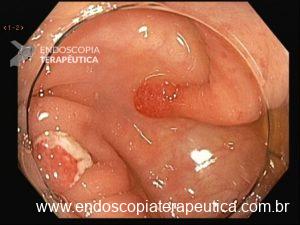
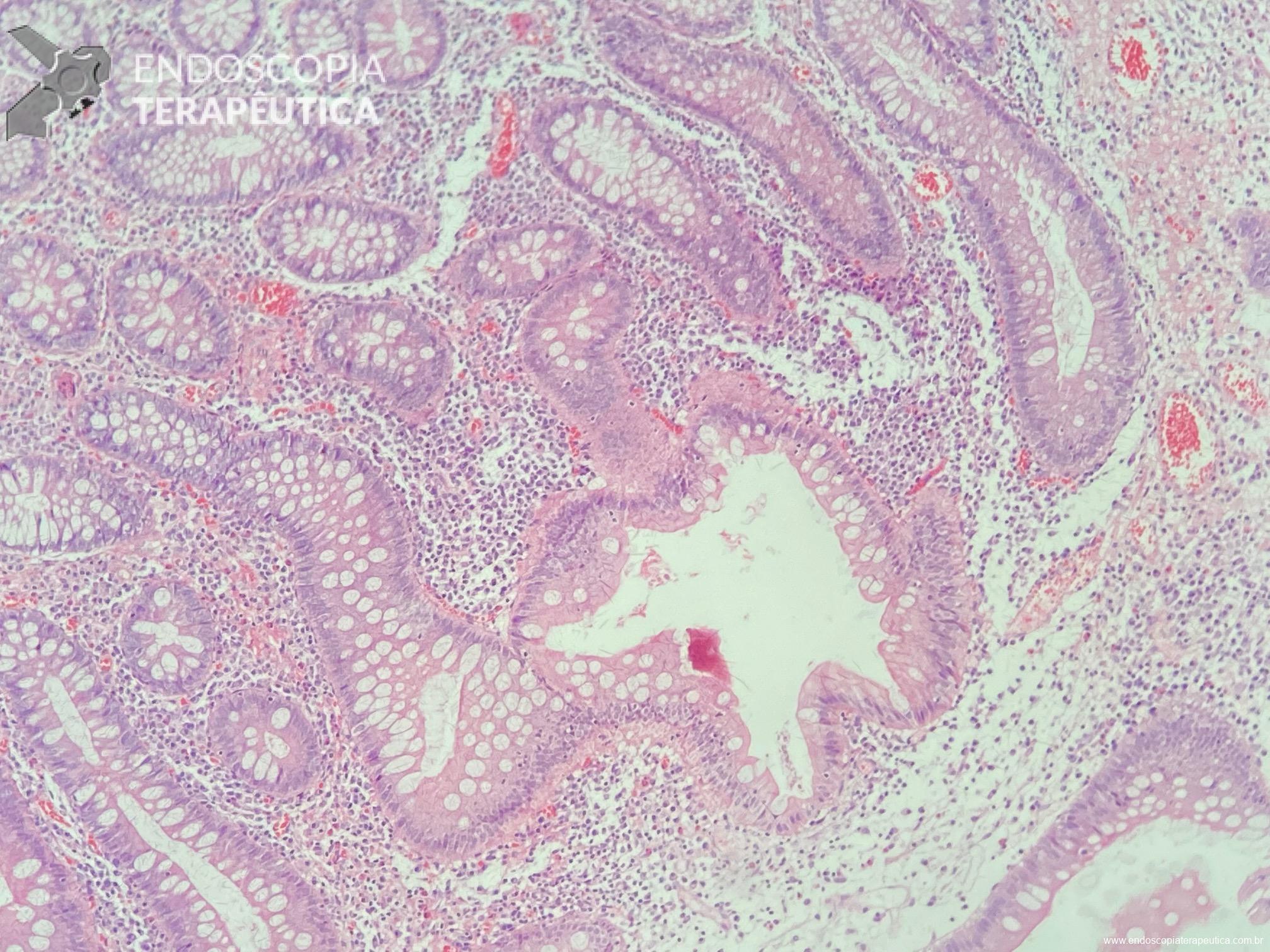
Será que UNDERWATER pode ser o futuro das ressecções de lesões de cólon?
O padrão atual para remoção de pólipos médios (10–20 mm) é Mucosectomia Convencional (EMR), que depende da injeção de solução salina para elevar a lesão. Porém, esse padrão-ouro tem uma falha importante: frequentemente não remove os tumores em bloco, levando a ressecções fragmentadas e taxas de recorrência entre 15% e 23,5%.
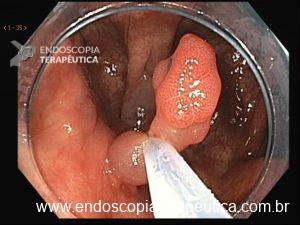
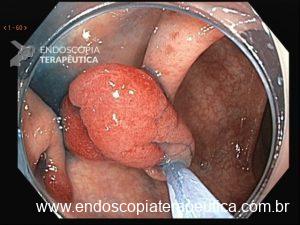
O segredo está na física. Ao injetar fluido sob o pólipo, criamos tensão na mucosa, o que dificulta ao endoscopista capturar profundamente a lesão com a alça. Recentemente, uma técnica vem revolucionando o campo: a Ressecção Endoscópica da Mucosa Subaquática, também conhecida como Mucosectomia Underwater (UEMR). Esta técnica elimina a injeção e preenche o cólon com água, permitindo que a flutuabilidade natural faça o trabalho.
Não é necessário injetar para alcançar profundidade
O achado mais surpreendente dos estudos clínicos recentes é que não é preciso um “colchão” salino para cortar com segurança e profundidade.
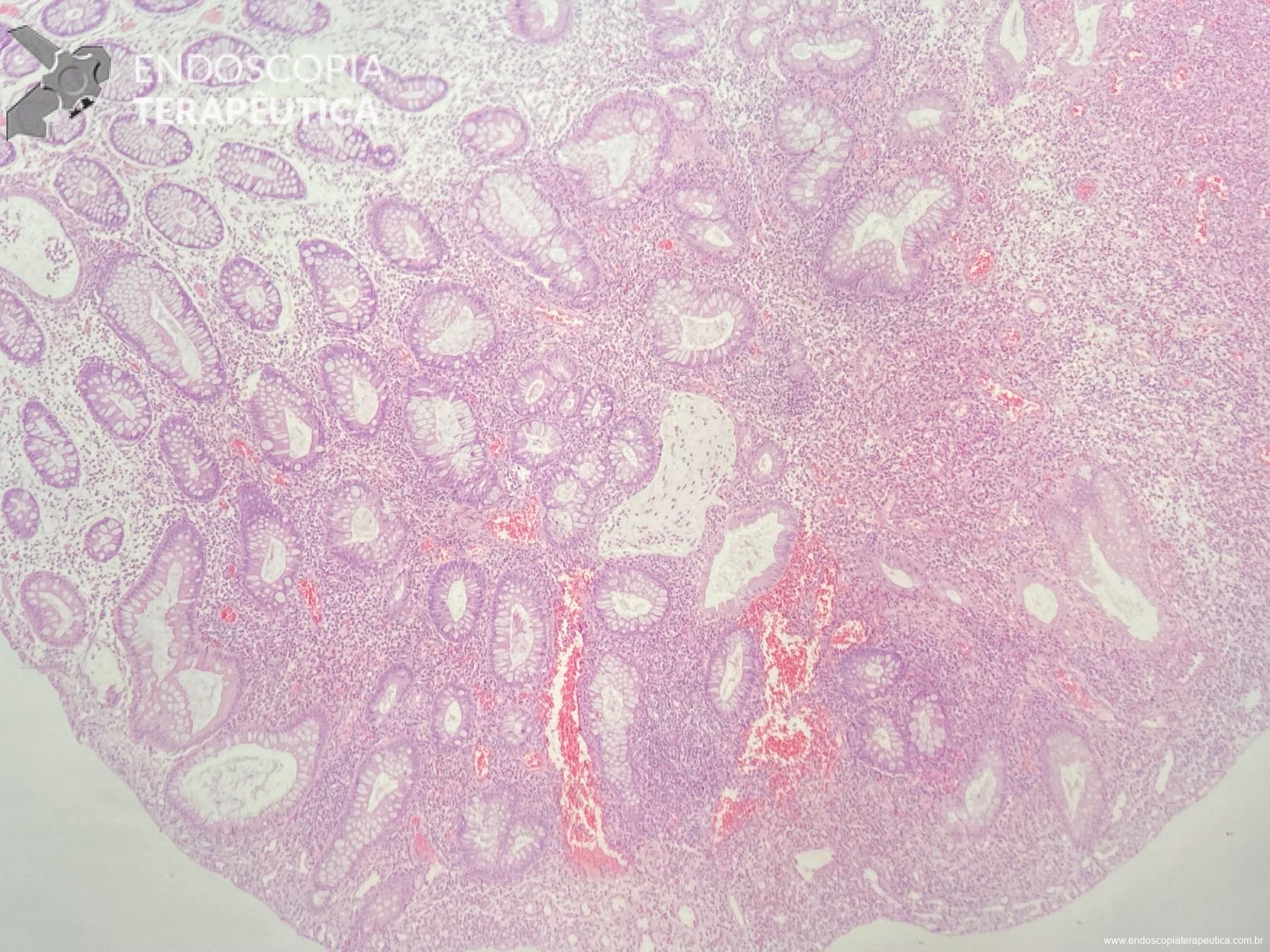
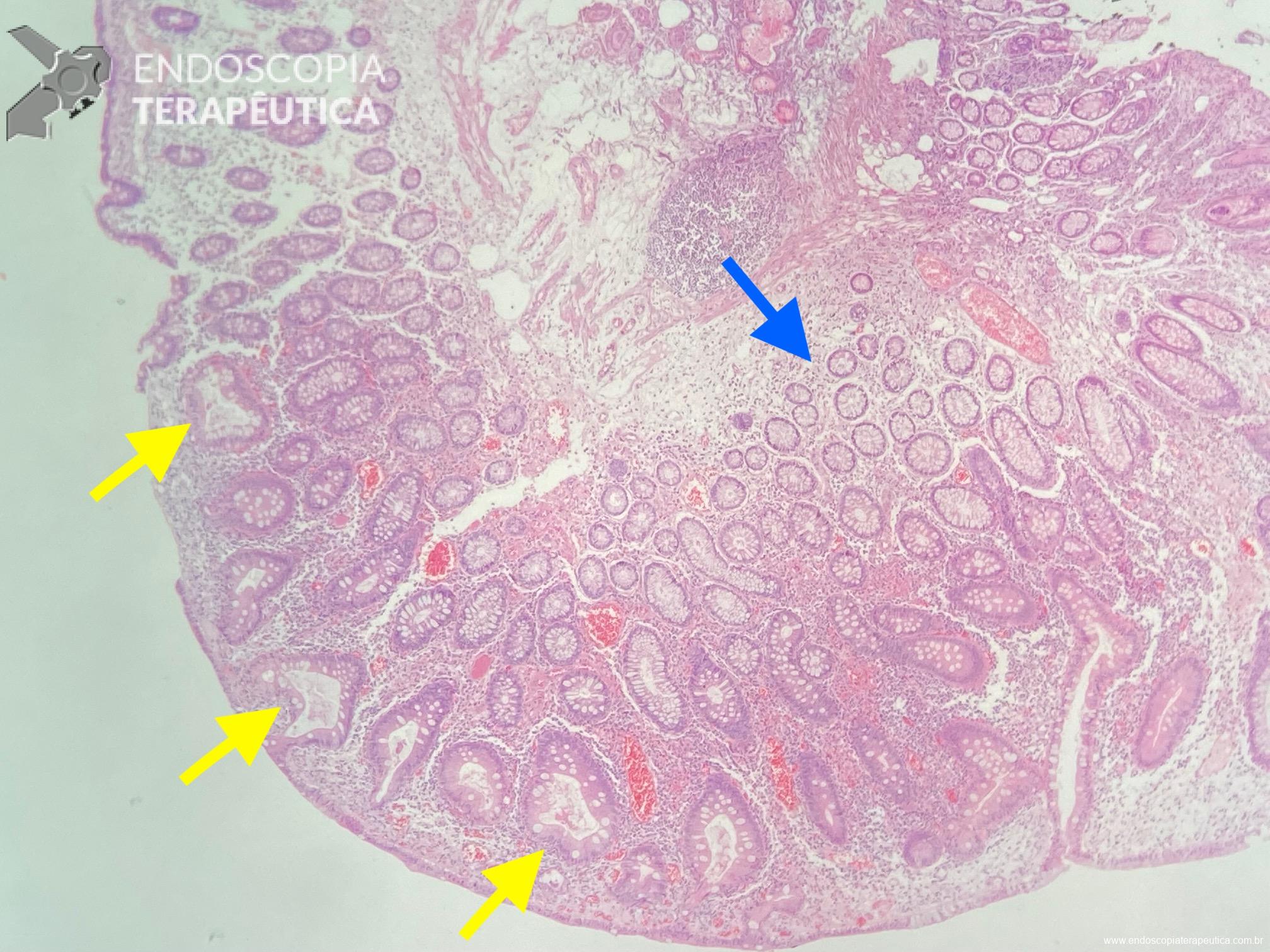
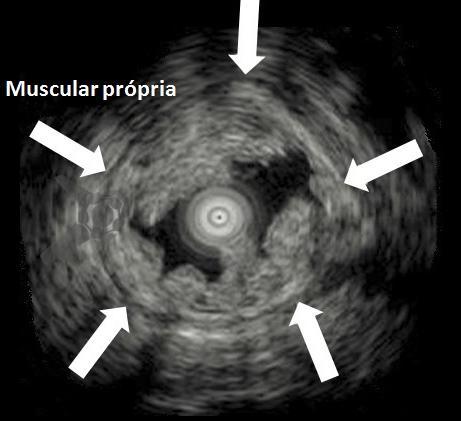
Em um estudo clínico randomizado, a UEMR atingiu profundidade média de 1.688,9 μm, numericamente maior que os 1.432,3 μm obtidos pelo método com injeção. Embora o resultado tenha mostrado apenas não inferioridade estatística (P=0,18), ele desmonta o mito de que a injeção é essencial para segurança.
O segredo é um fenômeno chamado redução gravitacional. Ao encher o cólon com água em vez de ar, a flutuabilidade faz a lesão “boiar” para longe da muscular própria, permitindo que a alça capture tecido submucoso adequado sem risco de envolvimento muscular ou perfuração.
UEMR e o cólon direito
O cólon direito, especialmente o ceco e o cólon ascendente são áreas de desafio para o endoscopista: paredes finas, curvas difíceis e maior dificuldade técnica. Mesmo assim, na UEMR, os resultados foram superiores: 1.822,4 μm de profundidade contra 1.096,5 μm com EMR (P=0,01).
A UEMR se destaca no cólon direito porque:
- as paredes finas tornam perigoso uma ressecção profunda com injeção,
- a água evita a tensão excessiva da insuflação com ar,
- há melhor manobrabilidade em ângulos difíceis.
Com água, a pressão intraluminal diminui e a parede não fica esticada, permitindo que o tumor protrua mais naturalmente e seja capturado com segurança.
Tratando pólipos planos
Pólipos superficiais (“flat”) são desafiadores porque costumam estar presos por fibrose submucosa, que impede a elevação com a injeção. A UEMR resolve isso: a flutuabilidade transforma a lesão plana em uma lesão polipoide, permitindo profundidades significativamente maiores (1.238,7 μm vs. 731,6 μm; P<0,01).
Debaixo d’água, a força de flutuação supera a fibrose e libera a base da lesão, facilitando a captura pela alça.
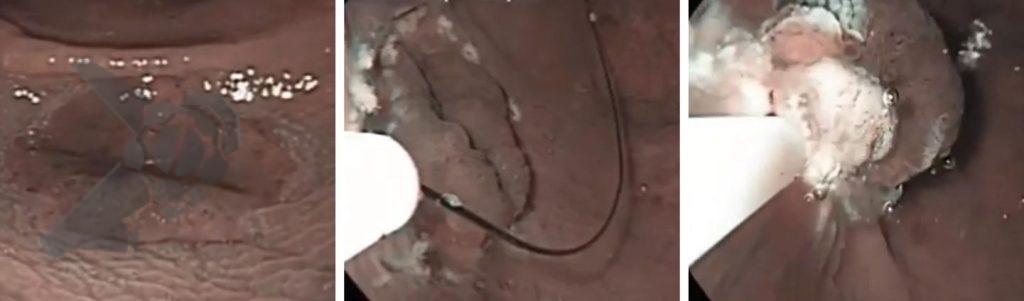
O grande equalizador dos endoscopistas
A descoberta mais impactante: a UEMR reduz a dependência da experiência do operador.
Endoscopistas com <10 anos de prática obtiveram profundidades maiores com UEMR (1.786,6 μm) do que com EMR (1.192,4 μm). Isso ocorre porque a injeção aumenta a tensão mucosa, tornando o alvo mais difícil. Já na UEMR, a lesão flutua, tornando o procedimento mais intuitivo e uniforme entre diferentes níveis de habilidade.
E tudo isso sem aumentar o tempo de procedimento (7,2 min vs. 6,5 min; P=0,34).
Conclusão: será que estamos em frente à um novo padrão-ouro?
Os dados mostram que a UEMR é:
- não inferior em profundidade total,
- superior no cólon direito,
- superior para lesões planas,
- mais estável para operadores menos experientes.
Enquanto aguardamos estudos internacionais maiores, o que já está claro é que estamos entrando em uma nova era no tratamento endoscópico das lesões do cólon, onde a ferramenta mais importante pode não ser uma agulha, mas sim os princípios básicos da física dos fluidos.
Veja mais sobre o tema:
- Mucosectomia underwater com auxílio de cap
- Ressecção de lesão extensa de cólon direito por UEMR
- Mucosectomia underwater de lesão de reto
Referência
- Tamaru Y, Miyakawa A, Shimura H, et al. Resection depth of underwater versus conventional endoscopic mucosal resection for intermediate-sized tumors. Am J Gastroenterol 2026 Jan 9.
Como citar este artigo
Orso IRB. Será que UNDERWATER pode ser o futuro das ressecções de lesões de cólon? Endoscopia Terapêutica 2026 Vol I. Disponível em: https://endoscopiaterapeutica.net/pt/assuntosgerais/sera-que-underwater-pode-ser-o-futuro-das-resseccoes-de-lesoes-de-colon/