Trata-se de uma forma incomum de pancreatite crônica que afeta a área localizada entre a parede duodenal medial, a cabeça do pâncreas e o colédoco (Figura 1). É uma causa pouco familiar para a maioria dos médicos e, portanto, geralmente subdiagnosticada [1]. Foi descrita pela primeira vez em 1970 por Potet e Duclert [2].
Muitos termos são considerados sinônimos de pancreatite paraduodenal (PP), como distrofia cística duodenal, cisto de parede paraduodenal, pancreatite de sulco (groove pancreatitis), mioadenomatose, distrofia cística do pâncreas heterotópico e também hamartoma duodenal pancreático [1,2,3,6].
Há duas formas de apresentação:
- pura: quando as alterações fibroinflamatórias acometem exclusivamente o sulco paraduodenal, com preservação do parênquima pancreático;
segmentar: quando há o acometimento da porção dorsomedial da cabeça do pâncreas, com acometimento do ducto pancreático principal (DPP).
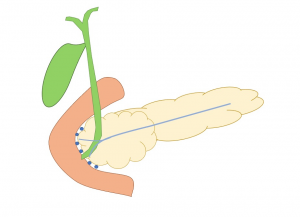
Figura 1: essa imagem mostra o sulco pancreáticoduodenal (círculos azuis), um “espaço teórico” entre a parede duodenal medial, cabeça do pâncreas e o colédoco.
Epidemiologia
Acomete majoritariamente homens caucasianos, com idade entre 40 e 50 anos, associado a histórico de alcoolismo pesado e, na maioria, concomitante a tabagismo de longa data [1,4]. A incidência não é bem estabelecida. Numa série de pacientes submetidos à pancreaticoduodenectomia por pancreatite crônica, a incidência variou de 2,7% a 24,5% [4].
Etiologia
É provavelmente heterogênea e multifatorial. Um mecanismo proposto sugere que o consumo de álcool e a exposição ao tabaco aumentam a viscosidade de secreções pancreáticas que levam à estase e à obstrução do ducto, ocasionando inflamação local crônica na papila menor e na área ao redor da cabeça do pâncreas, além de calcificações do ducto de Santorini. Fibrose e cicatriz resultam em estenose do colédoco e no aumento da rigidez da parede duodenal com posterior estenose [1,4].
Úlceras pépticas também são potenciais desencadeantes associadas à forma segmentar em 41% de casos na série de Solte et al. Outros fatores incluem hipersecreção gástrica, ressecções gástricas prévias, cistos verdadeiros da parede duodenal e da cabeça do pâncreas [3,6].
Apresentação clínica
Dor abdominal, perda de peso importante e mais raramente pródomos de obstrução digestiva alta. Quando há acometimento do DPP, pode ocorrer diarreia, diabetes mellitus e icterícia [1,2,3,4].
Laboratório
As enzimas pancreáticas e hepáticas podem estar ligeiramente elevadas. Os marcadores tumorais CEA e CA 19-9 geralmente são normais [3].
Apresentação microscópica
Os achados patológicos comuns consistem em lesões císticas tanto na submucosa duodenal quanto na muscular própria. Esses cistos podem conter fluidos claros, material granular espesso e, ocasionalmente, cálculos. As características histológicas mais comuns de PP incluem hiperplasia da glândula de Brunner, tecido pancreático heterotópico na submucosa ou na muscular própria da parede duodenal, proliferação miofibroblástica, células estromais fusiformes, macrófagos carregados de lipídios e detritos celulares granulares. Não há achado específico [3,4,10].
Exames de Imagens
Compressão extrínseca duodenal com mucosa sobrejacente aparentemente normal.
Tomografia computadorizada
Na forma pura, mostra uma massa laminar, em crescente, hipodensa, entre a cabeça pancreática e duodeno, perto da papila menor. A captação tardia de contraste é observada em alguns pacientes, devido ao fluxo sanguíneo reduzido causado pela proliferação de tecido fibrótico. [3]
Ressonância magnética (RM)
Massa laminar no sulco hipointensa em T1 em comparação com o parênquima pancreático. Na sequência T2, pode ser hipo-iso (quadro tardio) ou ligeiramente hiperintenso (caso agudo pelo edema). Não é frequente infiltração para o retroperitônio nem o acometimento de vasos [1,3].
Colangio-RM
É, atualmente, o principal recurso para visualizar o DPP e o ducto biliar comum. Revela um padrão regular, liso e suave de estreitamento da porção intrapancreática do colédoco distal, diferentemente dos casos de neoplasia [3].
Ecoendoscopia ± biópsia (EUS)
Ferramenta imprescindível na abordagem das lesões pancreaticobiliares. EUS localiza a anatomia exata e avalia a superfície envolvida, com a limitação que não é capaz de diferenciar infiltração e inflamação. Os achados da ecopunção podem mimetizar neoplasias, pela presença de células fusiformes abundantes ou grande número de células gigantes. Da mesma forma, área com fibrose não exclui neoplasia. Mas, em geral, EUS consegue diferenciar adenocarcinoma pancreático de PP, pela análise cito-histológica, em quase 90% dos casos [1,3,7,8,9].
Na série de Arvanitakis et al, o diagnóstico de PP foi baseado principalmente em RM e EUS, quando três sinais eram presentes:
- espessamento duodenal;
- cistos na parede duodenal;
- massa no sulco.
A presença desses três sinais demonstrou identificar corretamente a PP com uma especificidade de 88,2% [2].
Diagnóstico diferencial
Hamartona duodenal, tumores neuroendócrinos do sulco, principalmente, o gastrinoma, tumor estromal gastrointestinal, pancreatite autoimune, neoplasia mucinosa papilar intraductal, cisto de colédoco, colangiocarcinoma distal, carcinoma duodenal, divertículo periampular, metástase ampular e pancreatite aguda com necrose ou pseudocistos.
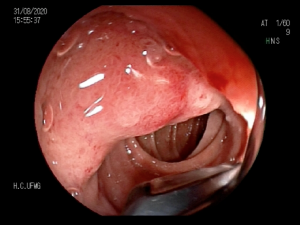
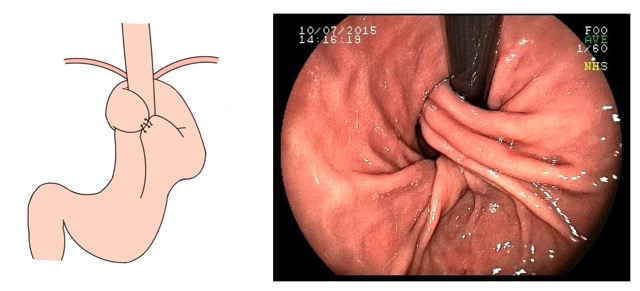
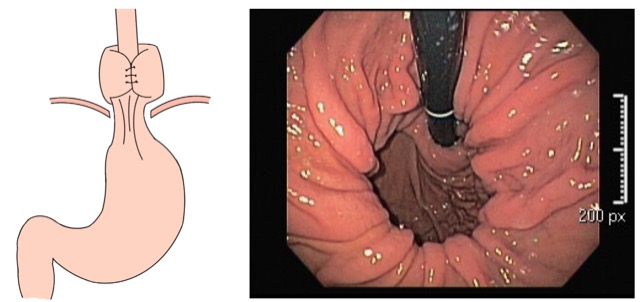
Imagem endoscópica mostra lesão com projeção endoluminal na segunda porção duodenal, proporcionando diminuição da luz do órgão. Imagens cedidas pelo Dr. Felipe A. Retes.
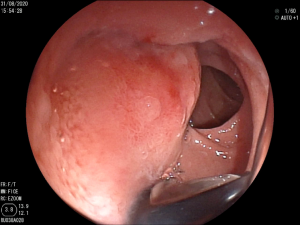
Imagem endoscópica mostra lesão com projeção endoluminal na segunda porção duodenal, proporcionando diminuição da luz do órgão. Imagens cedidas pelo Dr. Felipe A. Retes.
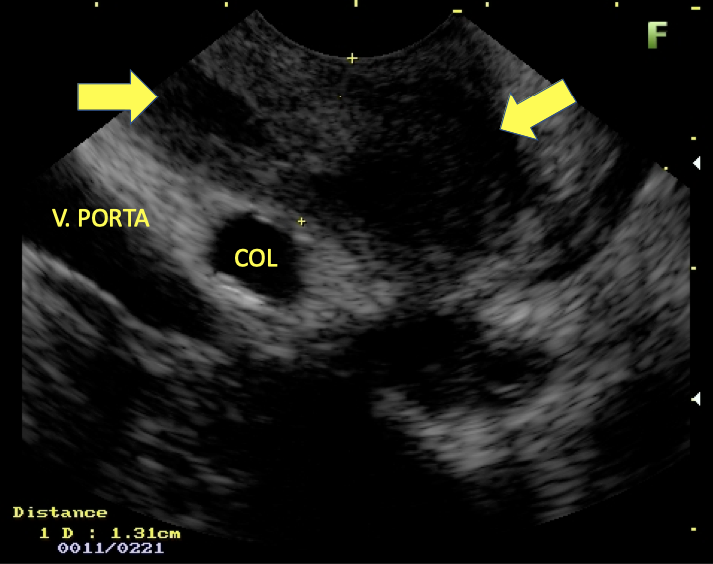
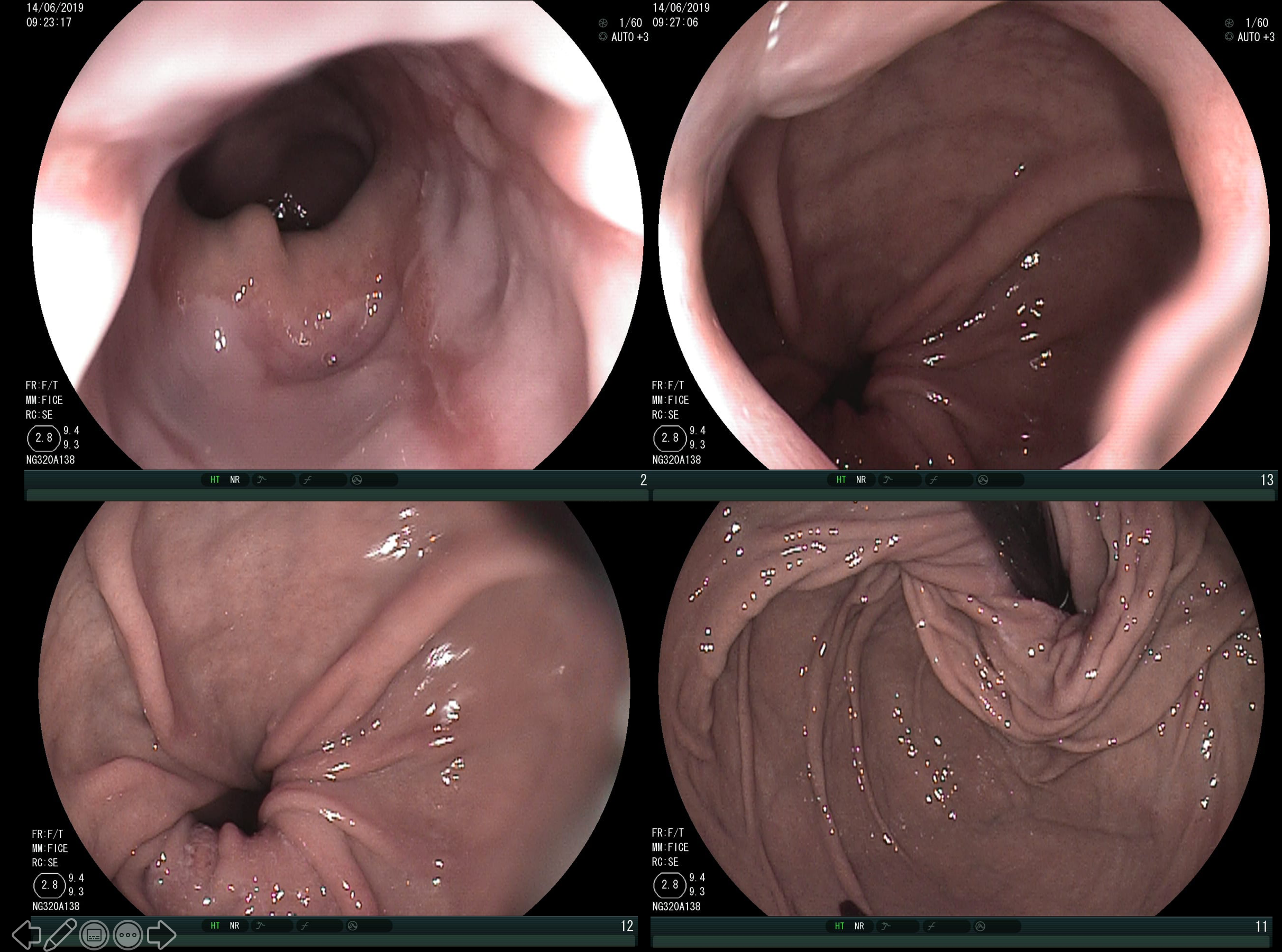
Imagem ecográfica demonstra espessamento da parede duodenal, medindo 1.31 cm de diâmetro, com áreas císticas de permeio (setas). Imagens cedidas pelo Dr. Felipe A. Retes.
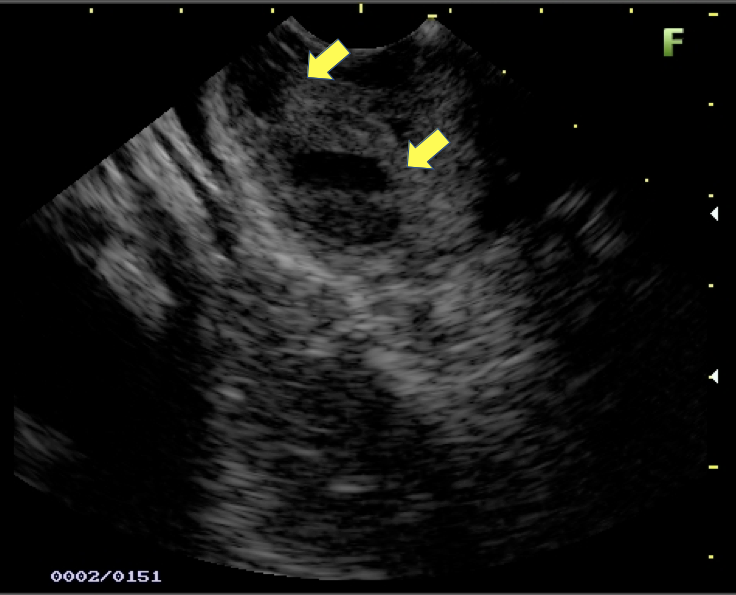
Imagem ecográfica demonstra espessamento da parede duodenal com áreas císticas de permeio (setas). Imagens cedidas pelo Dr. Felipe A. Retes.
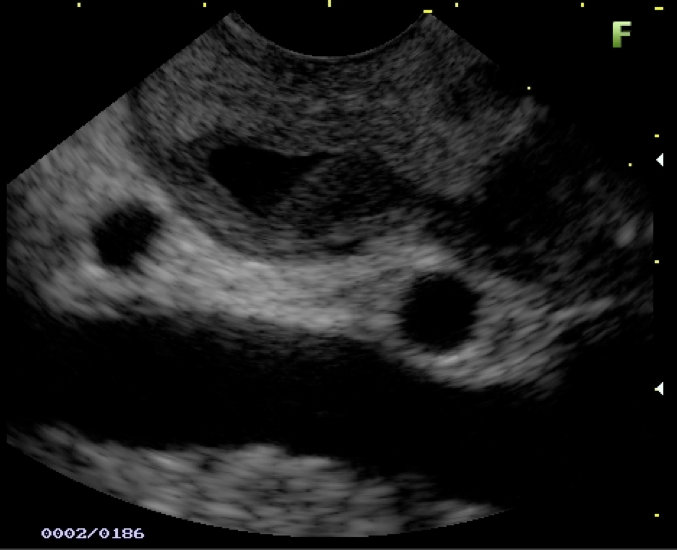
Imagem ecográfica demonstra espessamento da parede duodenal, com áreas císticas de permeio. Imagens cedidas pelo Dr. Felipe A. Retes.
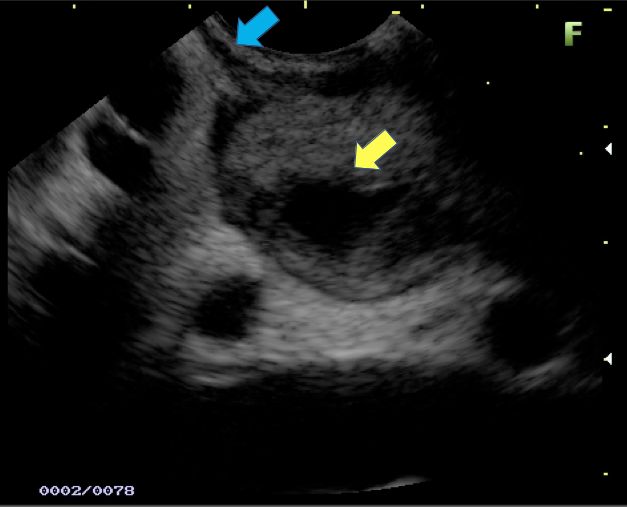
Transição da mucosa duodenal normal para área espessada (seta azul). Notam-se áreas císticas de permeio (seta amarela). Imagens cedidas pelo Dr. Felipe A. Retes.
Tratamento
Arvanitakis et al. mostrou que uma abordagem gradual para o tratamento da PP é viável, eficaz e está associada a uma taxa aceitável de complicações.
O tratamento dos sintomas agudos iniciais com medidas conservadoras (analgésicos, repouso pancreático e abstinência) pode ser útil a curto prazo. O uso da somatostatina demonstrou melhorar os resultados da abordagem não cirúrgica, sobretudo, na ausência de tratamento endoscópico devido à papila inacessível por compressão. Entre as desvantagens, está o alto risco de recorrência de sintomas após a interrupção do tratamento.
Em relação ao tratamento endoscópico, incluem-se várias modalidades, como drenagem do ducto pancreático, dilatação da estenose duodenal e drenagem endoscópica dos cistos.
Drenagem endoscópica através da papila menor melhora a dor e parece ser viável apenas no estágio inicial da doença, antes do desenvolvimento de cicatrizes graves e estenose duodenal importante.
A cirurgia é considerada o tratamento de escolha se sintomas refratários, nas complicações ou quando há suspeita de malignidade. A técnica de escolha é a duodenopancreatectomia cefálica ou a Técnica de Whipple [2,3,5].
Como citar este artigo
Araújo GAB. Você conhece a pancreatite paraduodenal (Groove pancreatitis)?. Endoscopia Terapêutica; 2021. Disponível em: https://endoscopiaterapeutica.net/pt/assuntosgerais/voce-conhece-a-pancreatite-paraduodenal-groove-pancreatitis
Referências
- Addeo G, Beccani D, Cozzi D, Ferrari R, Lanzetta MM, Paolantonio P, Pradella S, Miele V. Groove pancreatitis: a challenging imaging diagnosis. Gland Surg. 2019 Sep;8(Suppl 3):S178-S187. doi: 10.21037/gs.2019.04.06. PMID: 31559185; PMCID: PMC6755950.
- Arvanitakis M, Rigaux J, Toussaint E, Eisendrath P, Bali MA, Matos C, Demetter P, Loi P, Closset J, Deviere J, Delhaye M. Endotherapy for paraduodenal pancreatitis: a large retrospective case series. Endoscopy. 2014 Jul;46(7):580-7. doi: 10.1055/s-0034-1365719. Epub 2014 May 16. PMID: 24839187.
- Pallisera-Lloveras A, Ramia-Ángel JM, Vicens-Arbona C, Cifuentes-Rodenas A. Groove pancreatitis. Rev Esp Enferm Dig. 2015 May;107(5):280-8. PMID: 25952803.
- Larjani S, Bruckschwaiger VR, Stephens LA, James PD, Martel G, Mimeault R, Balaa FK, Bertens KA. Paraduodenal pancreatitis as an uncommon cause of gastric outlet obstruction: A case report and review of the literature. Int J Surg Case Rep. 2017;39:14-18. doi: 10.1016/j.ijscr.2017.07.043. Epub 2017 Jul 25. PMID: 28783521; PMCID: PMC5545816
- Kager LM, Lekkerkerker SJ, Arvanitakis M, Delhaye M, Fockens P, Boermeester MA, van Hooft JE, Besselink MG. Outcomes After Conservative, Endoscopic, and Surgical Treatment of Groove Pancreatitis: A Systematic Review. J Clin Gastroenterol. 2017 Sep;51(8):749-754. doi: 10.1097/MCG.0000000000000746. PMID: 27875360.
- Patel BN, Brooke Jeffrey R, Olcott EW, Zaheer A. Groove pancreatitis: a clinical and imaging overview. Abdom Radiol (NY). 2020 May;45(5):1439-1446. doi: 10.1007/s00261-019-02239-1. PMID: 31559471.
- Laugier R, Grandval P. Does paraduodenal pancreatitis systematically need surgery? Endoscopy. 2014 Jul;46(7):588-90. doi: 10.1055/s-0034-1377268. Epub 2014 Jun 30. PMID: 24979693.
- Ligresti D, Tacelli M, Amata M, Barresi L, Caruso S, Tarantino I, Traina M. Pure cystic groove pancreatitis: endosonographic appearance. Endoscopy. 2019 Aug;51(8):E235-E236. doi: 10.1055/a-0889-7569. Epub 2019 May 2. Erratum in: Endoscopy. 2019 Aug;51(8):C7. PMID: 31049896.
- Okasha H, Wahba M. EUS in the diagnosis of rare groove pancreatitis masquerading as malignancy. Gastrointest Endosc. 2020 Aug;92(2):427-428. doi: 10.1016/j.gie.2020.02.030. Epub 2020 Feb 27. PMID: 32112782.
- Jun JH, Lee SK, Kim SY, Cho DH, Song TJ, Park DH, Lee SS, Seo DW, Kim MH. Comparison between groove carcinoma and groove pancreatitis. Pancreatology. 2018 Oct;18(7):805-811. doi: 10.1016/j.pan.2018.08.013. Epub 2018 Aug 30. PMID: 30224296.
Acesse o Endoscopia Terapêutica para tomar contato com mais artigos comentados, assuntos gerais, casos clínicos, quizzes, classificações e mais!