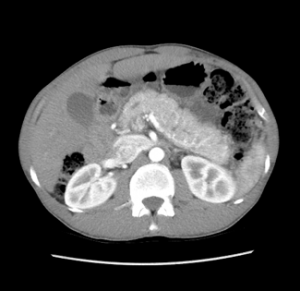
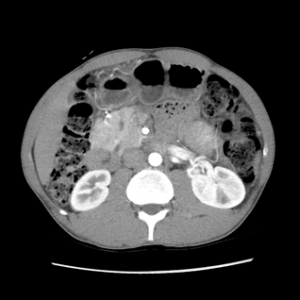
CISTOADENOMA SEROSO DE PÂNCREAS
Os cistos pancreáticos são, na maioria dos casos, achados incidentais de exames de imagem.
Estima-se que cerca de 3-14% das pessoas submetidas a exames abdominais tenham como achado alguma lesão cística pancreática. Em estudos de necropsia, esse achado pode chegar a 24%. Observa-se um claro aumento de prevalência em faixas etárias maiores.
As lesões císticas podem ser divididas em:
- cistos benignos: pseudocistos, cistos simples, cistoadenomas serosos
- cistos malignos: cistoadenocarcinomas, tumores neuroendócrinos císticos, neoplasia sólido-cística pseudopapilar
- cistos com potencial de malignização: IPMNs e cistoadenomas mucinosos
Nesse artigo falaremos um pouco sobre o cistoadenoma seroso.
CISTOADENOMA SEROSO (SCA)
O cistoadenoma seroso é uma lesão que acomete mais mulheres do que homens (2:1), na 6ª ou 7ª década de vida.
É uma lesão que não tem preferência por nenhuma região pancreática, podendo acometer cabeça, corpo ou cauda da glândula.
Aspecto radiológico
A característica mais marcante do cistoadenoma seroso é o achado de uma lesão policística, com septos fibrosos entre eles, formando um aspecto microcístico (70% dos SCA). Em cerca de 20-30% dos casos, os septos convergem para o centro da lesão, formando uma cicatriz central fibrosa (sinal mais típico do SCA). Em 20% dos casos observamos um aspecto de favo de mel, com múltiplos microcistos e septos finos entre eles.
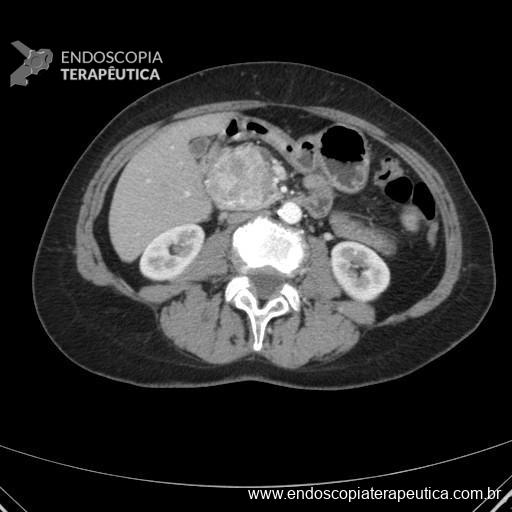
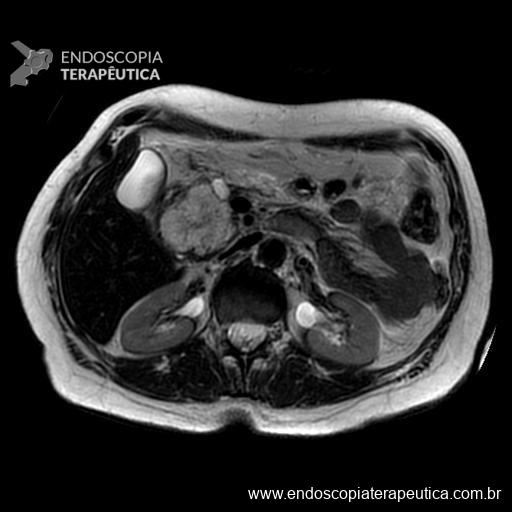
Figura 1: Cistoadenoma Seroso de cabeça de pâncreas – lesão lobular com septos convergindo para localização central da lesão. (arquivo pessoal)
Em cerca de apenas 10% dos casos os SCA podem ser oligocísticos, tornando o diagnóstico radiológico mais desafiador. Nesses casos, muitas vezes são necessários outros exames para confirmação diagnóstica, como a Ecoendoscopia com punção e análise do fluido intracístico.
Características do fluido
A característica citológica do cistoadenoma seroso são células cuboides, com citoplasma rico em glicogênio, embora a sensibilidade para citologia com PAAF seja muito baixa.
A análise bioquímica do líquido pode ajudar nos casos de diagnóstico incerto. A característica do SCA é ter o Antígeno Carcino-Embrionário (CEA) abaixo de 192 ng/ml, o que se associa a lesões não mucinosas. Além disso, por não haver comunicação com os ductos pancreáticos, a amilase no líquido intra-cístico é baixa.
Mais recentemente, com o avanço da endoscopia confocal, é possível a visualização do padrão de vascularização (nos SCA, ela é subepitelial – acurácia 87%) e permite biópsias do epitélio do cisto. Esse procedimento é realizado ainda em poucos centros, e embora melhore a acurácia do diagnóstico, traz maiores riscos de efeitos adversos (pancreatite aguda e hemorragia intracisto).
Prognóstico
O prognóstico dos SCA é excelente, com menos de 1% de mortalidade. Poucos casos na literatura tem evolução para malignidade, e não há uma concordância sobre a periodicidade do seguimento. Para muitos autores, trata-se de uma lesão benigna.
Embora seja uma lesão com baixa chance de transformação maligna, existe a possibilidade de crescimento da lesão em até 40% dos SCAs.
A última recomendação do grupo europeu é de um novo exame de imagem em 1 ano, e posteriormente, somente se houver sintomas (dor abdominal, icterícia ou náuseas e vômitos).
Referências
- Sakorafas, GH et al. Primary pancreatic cystic neoplasms revisited. Part I: Serous cystic neoplasms. Surgical Oncology, 2011
- Tirkes, T et al. Cystic neoplasms of the pancreas; findings on magnetic resonance imaging with pathological,surgical, and clinical correlation. Abdom Imaging, 2014
- Larson, A et al. Natural History of Pancreatic Cysts. Dig Dis Sci, 2017